Спиноцеребеллярная атаксия
OMIM 164400
Наша команда профессионалов ответит на ваши вопросы
В Центре Молекулярной Генетики проводится прямая молекулярно-генетическая диагностика наиболее частых форм спиноцеребеллярной атаксии: SCA 1, 2, 3, 6, 7, 8, 12 и 17 типов, которая основана на оценке числа CAG-повторов, локализованных в генах ATXN1, ATXN2, ATXN3, CACNA1A, ATXN7, ATXN8, PPP2R2B и TBP .
Спиноцеребеллярная атаксия 1 (SCA 1, OMIM 164400).
Заболевание обычно начинается в возрасте от 30 до 40 лет (возможный разброс – от 4 до 74 лет). Основные клинические симптомы – атаксия, офтальмоплегия, пирамидные и экстрапирамидные расстройства.
Молекулярно-генетической причиной SCA1 является увеличение числа тринуклеотидных САG повторов в гене ATXN1, располагающемся на 6-й хромосоме (сегмент 6р23). Длина гена составляет 450000 нуклеотидов. Ген содержит девять экзонов. Транскрипт состоит из 10660 нуклеотидов. В гене имеется участок тринуклеотидных повторов CAG (в норме их меньше 36; при болезни больше 40). Нормальные аллели гена обычно содержат в составе тринуклеотидного участка вставки от одного до трех CAT-триплетов, отсутствующие в мутантном гене. Их наличие рассматривается как важный фактор стабилизации нормальных аллелей при мейозе.
Спиноцеребеллярная атаксия 2 (SCA 2, OMIM 183090).
Спиноцеребеллярная атаксия 3 (SCA3, болезнь Мачадо-Джозефа, OMIM 109150)
Спиноцеребеллярная атаксия 6 (SCA6, OMIM 183086)
Спиноцеребеллярная атаксия 7 (SCA7, OMIM 164500)
Спиноцеребеллярная атаксия 7 типа (оливопонтоцеребеллярная атрофия 3 типа) – прогрессирующее аутосомно-доминантное нейродегенеративное заболевание, клинически характеризующееся церебеллярной атаксией, ассоциированной с дистрофией желтого пятна. Средний возраст манифестации заболевания – 32 года. Степень тяжести, скорость прогрессии и возраст начала заболевания варьируют как между семьями так и внутри семей. Основные клинические симптомы – офтальмоплегия, пирамидные и экстрапирамидные знаки, дизартрия, дисфагия, хорея, гиперрефлексия, спастика, потеря глубокой чувствительности, пигментная дегенерация сетчатки, прогрессирующая потеря зрения, медленные саккады, атрофия зрительного нерва.
Молекулярно-генетическая причина SCA7 –экспансия тринуклеотидных CAG-повторов гена ATXN7 (3p21.1-p12), находящихся в полиглутаминовом тракте белка ataxin-7.В норме количество повторов варьирует от 4 до 35, при болезни обнаруживается от 36 до 306 повторов.
Спиноцеребеллярная атаксия 8 (SCA8, OMIM 608768)
Спиноцеребеллярная атаксия 8 типа – медленно прогрессирующее аутосомно-доминантное заболевание, возраст начала которого варьирует от 18 до 65 лет. Основные клинические симптомы – прогрессирующая церебеллярная атаксия, нарушение координации походки, движения конечностей, речи, брадикинезия (замедленные движения). У больных часто наблюдается дизартрия, тремор, дисфагия, нистагм, замедление саккадических движений глазных яблок, дизметрические саккады, потеря чувствительности. При МРТ обнаруживается атрофия полушарий и червя мозжечка. Молекулярно-генетической причиной SCA8 является увеличение числа тринуклеотидных СAG повторов в гене ATXN8. В норме количество повторов варьирует от 15 до 50, при болезни обнаруживается от 71 до 1300.
Спиноцеребеллярная атаксия 12 (SCA12, OMIM 604326)
Молекулярно-генетическая причина SCA12 – увеличение числа тринуклеотидных CAG повторов в гене PPP2R2B, распологающемся на 5-ой хромосоме (сегмент q31-q33). В норме количество повторов от 7 до 32, при болезни обнаруживаются от 51 до 78. Клиническое значение экспансии повторов в диапазоне от 33 до 50 до сих пор не установлено. Ген PPP2R2B кодирует регуляторную субъединицу В белка фосфатазы 2 участвующего в таких регуляторных процессах, как рост и деление клеток, сокращение мышц и транскрипция генов.
Спиноцеребеллярная атаксия 17 (SCA 17, OMIM: 607136)
Молекулярно-генетической причиной SCA17 является увеличение числа тринуклеотидных CAG (или CAA) повторов в гене TBP, расположенном на длинном плече 6 хромосомы (сегмент q27). Ген TBP кодирует фактор транскрипции-белок, связывающий последовательность ТАТА-бокса (ТВР). В норме количество повторов варьирует от 25 до 44, при болезни обнаруживается от 47 до 63 повторов. Описана связь 45 и 46 повторов с неполной пенетрантностью заболевания.
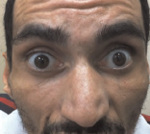
Болезнь Мачадо-Джозефа — генетически обусловленная спиноцеребеллярная атаксия, клинически представленная полиморфными сочетаниями мозжечкового синдрома с проявлениями вторичного паркинсонизма, гиперкинезами, пирамидными расстройствами в виде спастических параличей и офтальмоплегии, амиотрофиями. Диагностируется на основании тщательного изучения клинических проявлений у пациента и его родственников, генеалогического анализа, данных МРТ и КТ, выявления при исследовании ДНК превышающего норму количества копий триплета ЦАГ. Лечение симптоматическое. Прогноз неблагоприятный.
МКБ-10
Общие сведения
Болезнь Мачадо-Джозефа описана в средине 70-х годов 20 века. Предположительно, заболевание первоначально возникло у жителей Азорских островов, в связи с чем оно иногда встречается под названием «азорская болезнь». На сегодняшний день болезнь Мачадо-Джозефа распространена по всему миру. Ее случаи диагностированы у жителей США, Бразилии, Японии, Индии, Европы, Китая, Австралии, Канады. Она является наиболее встречаемой формой наследственной мозжечковой атаксии.
В рамках современной международной классификации заболеваний данная патология верифицируется как СЦА З — спиноцеребеллярная атаксия III тип. Характерна большая вариабельность времени дебюта (от 10 до 70 лет) и полиморфность клинических симптомов, обусловленная мультисистемным поражением церебральных и спинальных структур. В зависимости от комбинации основных клинических синдромов различают 3 варианта заболевания. Полиморфизм проявлений влечет за собой определенные затруднения в диагностике заболевания, которая может быть правильно реализована только при тесном сотрудничестве специалистов в области неврологии и генетики.
Причины болезни Мачадо-Джозефа
Ранее этиология была неизвестна. Благодаря развитию ДНК-исследований, выяснилось, что основным субстратом патологии выступает генная мутация, которая передается потомству аутосомно-доминантным путем. Аберрация локализуется в 14-й хромосоме (локус 14q24.3-q32) и заключается в экспансии (увеличении количества повторов) тринуклеотидного сочетания «цитозин-аденин-гуанин». Число повторов триплета ЦАГ существенно варьирует и в среднем составляет 62-84, в то время как в норме не превышает 37. Чем оно больше, тем ранее возникает манифестация болезни.
Морфологически наблюдается апоптоз нейронов зернистого слоя и клеток Пуркинье в коре мозжечка, дегенеративные изменения зубчатого и красного ядер, черной субстанции, моторных ядер черепно-мозговых нервов и передних рогов спинного мозга, спиномозжечковых трактов. В полосатом теле выявляются глиальные разрастания. Отличительной особенностью является интактность олив продолговатого мозга.
Клинические варианты болезни Мачадо-Джозефа
Болезнь Мачадо-Джозефа I тип манифестирует в возрастном периоде 10-30 лет. Характеризуется сочетанием пирамидных и экстрапирамидных симптомов. Пирамидный синдром (поражение кортикоспинальных трактов) обычно дебютирует спастическим парапарезом, затем присоединяется слабость в руках, парез мышц глотки с развитием дисфагии и дизартрии, парез глазодвигательных нервов с офтальмоплегией (симптом «фиксированных глазных яблок»). Наблюдается клонус стоп, патологические рефлексы. Экстрапирамидный синдром проявляется симптомами торсионной дистонии, атетозом, вторичным паркинсонизмом. Формируется скованная медленная походка с широкой расстановкой ног. Отмечается шаткость при ходьбе, обусловленная спастикой мышц, а не атаксией. Типичен экзофтальм, крупные фасцикулярные сокращения языка, не сопровождающиеся его атрофией. Возможны фасцикуляции мимических мышц, миокимия век. Наблюдается вертикальный и горизонтальный нистагм, саккады (однонаправленные движения глаз) с повышенной/пониженной амплитудой.
Болезнь Мачадо-Джозефа II тип дебютирует в период от 20 до 40 лет. Проявляется симптомами мозжечковой атаксии: абазией, гипер- и дисметрией, нарушением равновесия, дизартрией. Типично сочетание мозжечковой симптоматики с пирамидными и экстрапирамидными проявлениями, встречающимися при типе I. Офтальмоплегия и фасцикуляции наблюдаются гораздо реже, чем при болезни Мачадо-Джозефа I типа.
Болезнь Мачадо-Джозефа III тип представляет собой комбинацию мозжечковой атаксии и амиотрофий. Имеет наиболее позднее начало — после 40-летнего возраста. На фоне мозжечковых симптомов наблюдаются диффузные мышечные атрофии, сопровождающиеся гипотонией и слабостью, утратой сухожильных рефлексов. Отличительной особенностью выступает наличие расстройств всех видов чувствительности по дистальному типу, свидетельствующее о полиневропатии. Экзофтальм и лицевые фасцикуляции встречаются по различным данным у 20-50% пациентов с этой формой заболевания. Дегенерация кортикоспинальных трактов и поражение экстрапирамидной системы не характерны.
Диагностика болезни Мачадо-Джозефа
Большая вариабельность количества повторов триплета ЦАГ, наблюдаемая даже в пределах одной семьи, обуславливает значительный полиморфизм клинических проявлений болезни Мачадо-Джозефа, влекущий за собой существенные диагностические трудности. Распространенным является наличие различных типов заболевания у кровных родственников, особенно, если речь идет о разных поколениях. Описаны казуистические случаи, когда паркинсонизм выступал ведущим и единственным проявлением болезни. Таким образом, ключевым моментом в диагностике болезни Мачадо-Джозефа выступает консультация генетика, подробное исследование генеалогического древа с осмотром как можно большего числа родственников больного, проведение ДНК-диагностики.
С точки зрения невролога важным является выявление специфических отличий синдрома паркинсонизма, типичных для болезни Мачадо-Джозефа. Отсутствуют патогномоничные для болезни Паркинсона постуральные расстройства и тремор покоя, а неустойчивость в положении стоя и при ходьбе связана с статико-локомоторной атаксией. Паркинсонизм оказывается устойчивым к действию препаратов леводопы, хотя в начальных стадиях болезни может наблюдаться эффект в виде уменьшения мышечной ригидности.
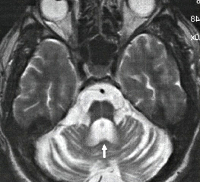
Первичная неврологическая диагностика (Эхо-ЭГ, ЭЭГ, РЭГ) не дает специфичных признаков, ее результаты могут быть в пределах нормы. КТ и МРТ головного мозга выявляет дегенерацию червя мозжечка и покрышки моста. Типичным признаком является выраженное расширение IV желудочка на фоне относительной сохранности мозжечковой коры. Дифференцируют болезнь Мачадо-Джозефа от других видов спиноцеребеллярных дегенерации, болезни Галлервордена-Шпатца, оливопонтоцеребеллярной дегенерации, атаксии Фридрейха, бокового амиотрофического склероза, атаксии Пьера-Мари, прогрессивного супрануклеарного паралича.
Лечение и прогноз болезни Мачадо-Джозефа
Эффективная терапия пока не найдена. Проводится симптоматическое лечение. При синдроме паркинсонизма показаны агонисты дофамина ( прамипексол, пирибедил), может применяться амантадин. Для снятия спастики назначаются фармпрепараты с миорелаксирующим действием (толперизон, баклофен). При гиперкинезах рекомендованы производные вальпроевой к-ты или бензодиазепины (клоназепам).
К сожалению, во многих случаях симптоматическая терапия оказывается не способной остановить прогрессирование болезни Мачадо-Джозефа. Наблюдается неуклонное усугубление симптоматики, приводящее к гибели пациента. Продолжительность жизни после дебюта заболевания составляет от 10 до 20 лет и зависит от клинического типа патологии. Наиболее скоротечным вариантом является болезнь Мачадо-Джозефа I типа. Профилактика заключается в проведении медико-генетического консультирования и пренатальной диагностики в отягощенных семьях; недопущении рождения ребенка, имеющего соответствующую генетическую мутацию.
Болезнь Мачадо-Джозефа
Болезнь Мачадо-Джозефа — довольно редкое неврологическое расстройство, проявляющееся в отсутствии мышечного контроля, что со временем при ненадлежащем лечении приводит к постепенному вырождению заднего мозга и гибели пациента.
Диагностика осуществляется посредством оценки клинических проявлений патологии как у самого больного, так и у его ближних родственников, проведении КТ и МРТ пациенту, а также оценки копии триплета ЦАГ.
К сожалению, для постановки правильного диагноза обойтись исключительно визитом к врачу не получится, так как только комплексная диагностика может выявить заболевание даже на начальной стадии, что предоставит возможность специалисту назначить своевременную терапию.
 Заболевание было впервые описано только в 70 гг. ХХ в. До этого периода в медицинской литературе нет никаких сведений о данной болезни. По предположениям ученых, патология впервые появилась у жителей Азорских островов, именно поэтому данное заболевание носит еще одно название — «азорская болезнь». Являясь самой распространенной формой мозжечковой атаксии, в наше время заболевание распространено повсеместно — случаи патологии были зарегистрированы в целом ряде стран.
Заболевание было впервые описано только в 70 гг. ХХ в. До этого периода в медицинской литературе нет никаких сведений о данной болезни. По предположениям ученых, патология впервые появилась у жителей Азорских островов, именно поэтому данное заболевание носит еще одно название — «азорская болезнь». Являясь самой распространенной формой мозжечковой атаксии, в наше время заболевание распространено повсеместно — случаи патологии были зарегистрированы в целом ряде стран.
Синдром Мачадо-Джозефа может проявить первые признаки у людей с 10 до 70 лет — довольно широкий возрастной диапазон, во время которого заболевание проявляет себя. Вследствие мультисистемного поражения спинальных и церебральных структур, проявления патологического процесса могут быть самыми разнообразными, что очень часто становится причиной постановки неправильного диагноза.
В зависимости от выраженности синдромов и характерных жалоб пациентов, принято различать три типа патологии. Множество форм проявлений очень сильно затрудняет диагностические мероприятия. Диагностика может быть осуществлена при совместной работе генетиков и неврологов. Только комплексная диагностика, проведенная при помощи новейших диагностических методик, которая будет оцениваться различными специалистами, способна предоставить максимально развернутую информацию, на основании которой будет поставлен правильный диагноз.
Причины
 В тот период, когда патология была впервые обнаружена, специалисты не могли дать однозначного ответа на вопрос о том, каковы причины ее возникновения. Даже несмотря на то, что было выдвинуто множество предположений, ни одно из них не нашло своего научного подтверждения.
В тот период, когда патология была впервые обнаружена, специалисты не могли дать однозначного ответа на вопрос о том, каковы причины ее возникновения. Даже несмотря на то, что было выдвинуто множество предположений, ни одно из них не нашло своего научного подтверждения.
Однако, с течением времени и по мере развития ДНК-исследований стало ясно, что главными причинами болезни выступает мутация генов, передаваемая по наследству аутосомно-доминантным путем.
Таким образом удалось выяснить, что заболевание имеет наследственный характер. Отклонения от нормы наблюдаются в 14 хромосоме и сводятся к экспансии комбинации «цитозин-аденин-гуанин». Количество повторов триплета ЦАГ очень сильно различается, в среднем показатели держатся в диапазоне от 62 до 84. Для сравнения: в норме эта цифра не должна быть выше 37. Чем данный показатель выше, тем раньше возникает болезнь у человека.
У больных возникает апоптоз нейронов в коре головного мозга, дегенеративные процессы красного и зубчатого ядер, моторных ядер ЧМ-нервов. Поражение зон мозга приводит к возникновению целого ряда признаков, которые по мере развития патологии и отсутствия соответствующего лечения начинают тревожить больного. Специалисты сходятся во мнении, что от момента возникновения патологии до проявления первых ее симптомов проходит от 3 до 6 месяцев, но многое зависит также от индивидуальных особенностей организма – у одних больных патология может проявиться значительно раньше, у других – позже.
Клинические варианты патологии
 Патология первого типа возникает в возрастном диапазоне 10-30 лет. У больных ярко выражены экстрапирамидные и пирамидные симптомы развития патологии. Пирамидный синдром возникает вместе с спастическим парапарезом, постепенно появляется значительная слабость в верхних конечностях, парез мышц глотки, глазодвигательных нервов, в результате чего человек теряет способность передвигать глазными яблоками.
Патология первого типа возникает в возрастном диапазоне 10-30 лет. У больных ярко выражены экстрапирамидные и пирамидные симптомы развития патологии. Пирамидный синдром возникает вместе с спастическим парапарезом, постепенно появляется значительная слабость в верхних конечностях, парез мышц глотки, глазодвигательных нервов, в результате чего человек теряет способность передвигать глазными яблоками.
Со временем развиваются патологические рефлексы, клонус стоп. Больные начинают испытывать трудности при передвижении, походка становится неуклюжей, скованной, при этом человек ходит с широко расставленными ногами. Очень часто такие пациенты сильно шатаются во время ходьбы. Нередко у больных смещаются глазные яблоки вперед, возникают значительные фасцикулярные сокращения языка, мимических мышц, глазных век. Имеют место быть непроизвольные движения глаз высокой частоты.
Патология второго типа возникает чаще всего с 20 до 40 лет. Симптомы заболевания проявляются в мозжечковой атаксии: возникает дизартрия, больной не способен сохранять равновесие, появляется абазия. Характерной комбинации мозжечковых и пирамидных признаков, но второй тип имеет некую схожесть с первым типом болезни.
Патология третьего типа выражается в комбинации амиотрофий и мозжечковой атаксии. Данный тип заболевания возникает довольно поздно – как правило, только после 40 лет. У больных появляется мышечная атрофия, которая проявляется сильной слабостью и полной потерей сухожильных рефлексов. Особенностью данного типа болезни является наличие всех видов расстройств чувствительности по дистальному типу. Дегенеративные изменения в кортикоспинальных трактах и поражение экстрапирамидной системы не наблюдается.
Диагностика
 Диагностика болезни осложняется тем, что ей свойственна огромная вариабельность повторов триплета ЦАГ, в результате чего болезнь проявляется в самых разнообразных симптомах даже в том случае, если возникает у членов одной семьи. Очень часто болезнь поражает разные поколения близких родственников, причем не обязательно проявляется в одном и том же типе.
Диагностика болезни осложняется тем, что ей свойственна огромная вариабельность повторов триплета ЦАГ, в результате чего болезнь проявляется в самых разнообразных симптомах даже в том случае, если возникает у членов одной семьи. Очень часто болезнь поражает разные поколения близких родственников, причем не обязательно проявляется в одном и том же типе.
Известны случаи, когда только одно из проявлений позволяло заподозрить наличие болезни у разных людей, имеющих между собой родственную связь. Одним из важнейших моментов в диагностике заболевания является консультация генетика, который осуществляет исследование генеалогического древа, учитывая большое количество родственников, а также проводя ДНК-исследование.
Немаловажным является и посещение невролога. Врач проведет первичный осмотр, по результатам которого выявит отличия болезни от других заболеваний.
К сожалению, в некоторых случаях осмотр у невролога не позволяет выявить характерные признаки заболевания из-за стертой клинической картины, поэтому врач может только заподозрить патологию, но поставить такой диагноз без более подробного обследования не получится. Пациентов с подозрениями на данный недуг в обязательном порядке направляют на КТ и МРТ, а после чего врач-рентгенолог определит наличие изменений в головном мозге. Важно понимать, что только обследование, проведенное на современных аппаратах, предоставит возможность врачам дать оценку возникшим изменениям в структуре головного мозга, на основании которых будет поставлен правильный диагноз.
Лечение и прогноз
 К сожалению, в настоящее время заболевание считается неизлечимым и никакого специального лечения, направленного на остановку дегенеративных процессов пока разработать не удалось. Поэтому все мероприятия направлены исключительно на снятие симптомов и улучшение качества жизни больного. При своевременно начатом лечении, которое было подобрано верно, а также соблюдении всех рекомендаций врачей, больные получают все шансы на замедление дегенеративных процессов, происходящих на фоне протекания болезни.
К сожалению, в настоящее время заболевание считается неизлечимым и никакого специального лечения, направленного на остановку дегенеративных процессов пока разработать не удалось. Поэтому все мероприятия направлены исключительно на снятие симптомов и улучшение качества жизни больного. При своевременно начатом лечении, которое было подобрано верно, а также соблюдении всех рекомендаций врачей, больные получают все шансы на замедление дегенеративных процессов, происходящих на фоне протекания болезни.
Однако, полного выздоровления не наступает даже при правильной терапевтической тактике. Если имеет место быть синдром паркинсонизма, назначают агонисты дофамина, иногда пациентам показано применение амантадина. С целью снятия спастики выписываются лекарственные препараты с миорелаксирующим действием.
Как уже говорилось выше, почти всегда даже хорошее лечение не сможет остановить дальнейшее развитие заболевания, поэтому с течением времени у больных появляются новые симптомы патологии, что в конечном итоге приводит к смерти. Согласно официальным статистическим данным, после выявления заболевания до гибели больного проходит от 10 до 20 лет.
Продолжительность жизни имеет прямую связь с типом заболевания. Неблагоприятный прогноз имеет первый тип Масадо-Джозефа, больные с таким диагнозом, как правило, не живут дольше 10 лет после начала прогрессирования патологии.
Профилактические мероприятия сводятся к проведению пренатальной диагностики, а также в получении консультации у генетиков. Если у ближайших родственников был диагностирован синдром Мачадо-Джозефа, важно со всей ответственностью подходить к планированию и рождению ребенка, так как генная мутация может передаться по наследству. Многие генетики сходятся во мнении, что очень важно не допускать рождение ребенка, у которого имеется характерная генетическая мутация.
Болезнь Мачадо – Джозефа (MJD), также известен как Азорская болезнь Мачадо – Джозефа, Болезнь Мачадо, Болезнь Иосифа или спиноцеребеллярная атаксия 3 типа (SCA3), является редким аутосомно-доминантно унаследованный нейродегенеративный болезнь, вызывающая прогрессирующее мозжечок атаксия, [1] [2] что приводит к отсутствию мышечный контроль и координация из верхний и нижние конечности. [3] Симптомы вызваны генетическим мутация что приводит к увеличению количества аномальных тринуклеотидных повторов «CAG» в ATXN3 ген [1] что приводит к аномальной форме белка атаксин что вызывает дегенерацию клеток в задний мозг. [3] Из-за некоторых симптомов, таких как неуклюжесть и жесткость, MJD обычно принимают за пьянство или болезнь Паркинсона.
Содержание
Симптомы
Симптомами MJD являются нарушения памяти, [4] спастичность, трудности с речью и глотанием, слабость в руках и ногах, неуклюжесть, частое мочеиспускание и непроизвольные движения глаз. Симптомы могут начаться в раннем подростковом возрасте и со временем ухудшаются. В конце концов, MJD приводит к параличу; однако интеллектуальные функции обычно остаются прежними. [ нужна цитата ]
Генетика
Флорес и Сан-Мигель являются очагами болезни Мачадо – Джозефа в Азорские острова. [5]
Болезнь Мачадо Джозефа имеет множественное происхождение, поскольку SCA3 происходит от гаплотипа четырех разных источников, а не от одного происхождения на Азорских островах. Япония, Бразилия и Франция все они были эффектами основателя в областях с SCA3. [6]
В Китаймутация, вызывающая SCA типа 3, произошла от 8000 до 17000 лет назад. [11]
В Японии самая старая причинная мутация произошла около 5774 +/- 1116 лет назад. [12]
Среди австралийских аборигенов мутация-основатель, по-видимому, произошла около 7000 лет назад. Поскольку эта мутация характерна для других семей, живущих в Азии, вполне вероятно, что она была импортирована в Австралию. [13]
MJD затронул азорцев, и у японцев были общие гаплотипы, в то время как у азорцев также были гаплотипы двух разных источников. [14]
Патофизиология
В мосты (сооружение, расположенное на мозговой ствол) является одной из областей, затронутых MJD. В полосатое тело (область мозга, связанная с балансом и движением) также поражена этим заболеванием, что может объяснить как основные двигательные проблемы, вызываемые MJD: сжатие и скручивание конечностей, так и резкие, нерегулярные движения. [16]
В пораженных клетках этот белок накапливается и собирает внутриядерные тельца включения. Предполагается, что эти нерастворимые агрегаты нарушают нормальную активность ядра и вызывают дегенерацию и гибель клетки. [ нужна цитата ]
Диагностика
MJD можно диагностировать, распознав симптомы болезни и взяв семейный анамнез. Врачи задают пациентам вопросы о типах симптомов, которые были у родственников с заболеванием, о прогрессировании и резкости симптомов, а также о возрасте начала заболевания у членов семьи. [ нужна цитата ]
Пресимптоматический диагноз MJD может быть установлен с помощью генетического теста. [17] Прямое обнаружение генетической мутации, ответственной за MJD, доступно с 1995 года. [18] Генетическое тестирование проверяет количество повторов CAG в кодирующей области гена MJD / ATXN3 на хромосоме 14. Тест покажет положительный результат на MJD, если эта область содержит 61-87 повторов, в отличие от 12-44 повторов, обнаруженных у здоровых людей. лиц. Ограничение этого теста заключается в том, что если количество повторов CAG у испытуемого человека находится между нормальным и патогенным диапазоном (45-60 повторов), то тест не может предсказать, будут ли у человека симптомы MJD. [17]
Классификация
Есть пять подтипов MJD. [1] которые характеризуются возрастом начала и диапазоном симптомов. Подтипы иллюстрируют широкий спектр симптомов, которые могут испытывать пациенты. [1] Однако отнесение людей к конкретному подтипу заболевания имеет ограниченное клиническое значение. [1]
лечение
Нет лекарства от болезни Мачадо-Джозефа. Однако для некоторых симптомов доступны методы лечения. [1] [3] Например, спастичность можно уменьшить с помощью спазмолитических препаратов, таких как баклофен. Симптомы паркинсонизма можно лечить с помощью леводопа терапия. Призменные очки могут уменьшить диплопические симптомы. [3] Физиотерапия/Физиотерапия и / или трудотерапия может помочь пациентам, выписав средства передвижения повысить независимость пациентов, обеспечивая тренировка походки, и прописывать упражнения для поддержания подвижности различных суставов и общего состояния здоровья, чтобы снизить вероятность падений или травм в результате падений. Ходунки и инвалидные коляски могут очень помочь пациенту в повседневных задачах. Некоторые пациенты будут испытывать трудности с речь и глотание, поэтому Речевой патолог может помочь пациентам улучшить свои коммуникативные способности и проблемы с глотанием. [3]
Прогноз
Ожидаемая продолжительность жизни пациентов с тяжелыми формами MJD составляет около 35 лет. Те, у кого легкие формы, имеют нормальную продолжительность жизни. Причиной смерти тех, кто умирает рано, часто является аспирационная пневмония. [3]
История
Заболевание впервые было выявлено в 1972 году. [19]
В отличие от многих других заболеваний, болезнь Мачадо – Джозефа не названа в честь исследователей. Он назван в честь двух мужчин («Уильям Мачадо» и «Антон Джозеф»), которые были патриархами семей, в которых изначально было описано состояние. [20] Самый высокий показатель распространенности этого заболевания в Австралии. Грут Эйландт где 5% населения в настоящее время имеют симптомы или находятся в группе риска, [21] за которым следует азорский остров Флорес где примерно 1 из 140 человек в популяции имеет диагноз MJD. [22]
Известные случаи
Бразильский комик, актер и телеведущий Гильерме Карам была диагностирована болезнь Мачадо – Иосифа, унаследованная от матери, как и его брат и сестра. [23] Он умер 7 июля 2016 года. [24] В видео на Facebook через несколько дней после смерти Карама бразильский деятель, журналист и телеведущий, Арнальдо Дюран публично признал свою болезнь Мачадо-Джозеф. Бразильская пресса уделила огромное внимание обоим случаям. [25]
Этическое соображение
Специалисты по этике использовали болезнь Мачадо – Джозефа как парадигматическую болезнь, чтобы обсудить права сообщества пациентов контролировать «право собственности» на свое заболевание, особенно когда речь идет об исследованиях генетического тестирования. [26] Кроме того, поскольку в настоящее время не существует клинического вмешательства для предотвращения появления симптомов заболевания, существует дискуссия о том, следует ли людям проходить тестирование или нет. [18] Преимущества тестирования MJD включают уменьшение беспокойства и неуверенности, а также способность планировать будущее. К некоторым недостаткам можно отнести ожидание отрицательных результатов и трудности человека в адаптации к этому результату. [ нужна цитата ]
Этнографический пример изучения некоторых социальных и этических последствий жизни с болезнью Мачадо – Джозефа см. В João Biehl’s Vita: Жизнь в зоне отказа от общества (Беркли: Калифорнийский университет Press, 2006).


